2024-05-05 Sunday

Time:2020-03-17 Reading:9727
Hydroformylation is one of the most widely used large-scale homogeneous catalytic reactions in today's chemical industry, converting olefins, hydrogen, and carbon monoxide into aldehydes and related chemicals with an annual production exceeding 10 million tons. Over the years, traditional industrial catalyst systems have shown excellent catalytic performance but have also faced significant challenges.
The first hydroformylation catalyst in history was the cobalt complex HCo(CO)4, accidentally discovered by Otto Roelen in 1938. This catalyst required high pressure, and the carbon monoxide partial pressure needed to increase rapidly with temperature to suppress catalyst decomposition. Subsequently developed phosphine-ligated cobalt catalysts HCo(CO)3(PR3) could catalyze the reaction at lower pressures. However, the strong Co-Co bond greatly influenced the catalytic rate, requiring higher reaction temperatures and ultra-high catalyst concentrations, which also led to more side reactions.
In the early 1970s, rhodium catalysts with hundreds of times higher activity than cobalt catalysts were introduced and later became the industrial standard rhodium-phosphine catalyst HRh(CO)(PPh3)2. Not only did this catalyst exhibit high activity, but it also operated at considerably lower pressures. However, this catalyst had its drawbacks. Apart from the high cost and low reserves of rhodium, the PPh3 ligand tended to dissociate, necessitating the use of excess PPh3 to maintain catalyst performance.
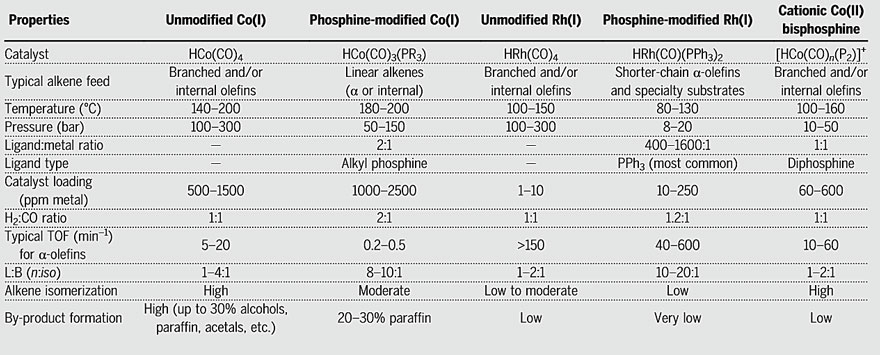
Professor George G. Stanley's research group at Louisiana State University in the United States has reported a highly active and selective cationic rhodium catalyst containing a tetraphosphine ligand, where the phosphine ligands bridge and chelate two rhodium centers (Science, 1993, 260, 1784–1788; J. Am. Chem. Soc., 2003, 125, 11180–11181). This catalyst exhibits excellent activity and selectivity for the hydroformylation of 1-hexene, but unfortunately, it suffers from degradation and deactivation issues. Recently, the research group shifted their focus back to the original cobalt catalyst. Unlike the previous neutral cobalt(I) complex catalysts, their newly developed cationic cobalt(II) bisphosphine complex ([HCo(CO)n(P2)]+) shows higher activity at lower pressures, approaching the activity of currently used industrial precious metal rhodium catalysts. This positively charged hydrogenation catalyst offers several advantages: (1) high reactivity and fast reaction rate, capable of catalyzing the hydroformylation of internally branched alkenes; (2) does not require high pressure or excessively high temperatures; (3) more stable and does not require excess ligands or catalysts; (4) higher turnover frequency (TOF) with fewer side reactions; and (5) significantly lower cost compared to rhodium catalysts. These findings have been published in Science.

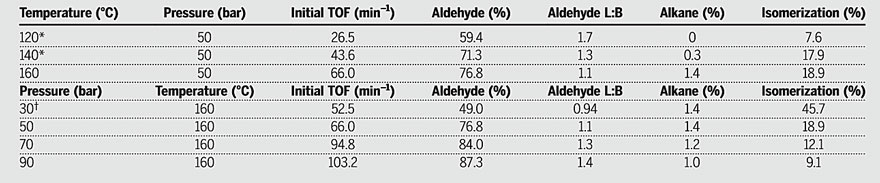
Under mild conditions, the hydrogenation activity of the monometallic catalyst precursor [Co(acac)(DPPBz)](BF4) is higher than that of the dicobalt complex. Figure 3 illustrates the effect of temperature and pressure on the catalytic performance of this catalyst for the hydroformylation of 1-hexene. Under 50 bar H2:CO (1:1), as the temperature increases, the catalyst's activity increases, but at 170 °C, the catalyst begins to decompose. To address this, the authors increased the H2:CO pressure at 160 °C and found that the activity gradually increased. The initial turnover frequency (TOF) increased from 52.5 min-1 (30 bar) to 103.2 min-1 (90 bar). This cationic Co(II) catalyst exhibits high alkene isomerization activity similar to the Co(I) catalyst system. The low linear/branched (L:B) selectivity of aldehyde products suggests that the cationic cobalt catalyst can coordinate with internally formed alkenes during isomerization and subsequently hydroformylate them.。
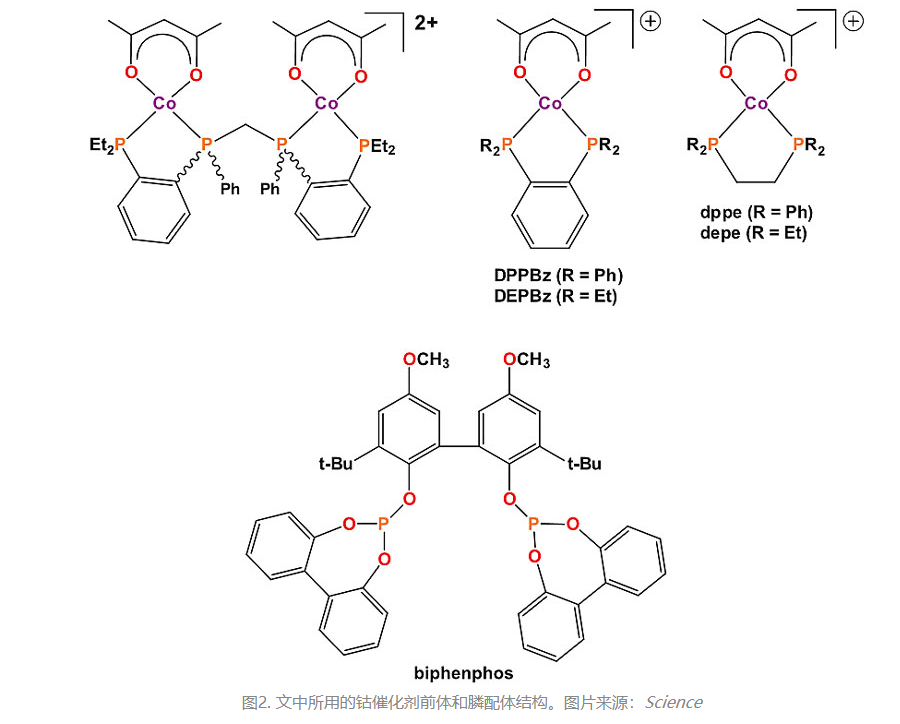
The use of electron-rich bisphosphine ligands, such as depe, allows for the synthesis of cationic Co(II) hydrogenation catalysts with higher activity at low to moderate pressures. Under standard conditions, the catalyst [Co(acac)(depe)](BF4) is activated at 140 °C and 34 bar H2:CO (1:1) for 5 minutes. Then, the temperature and pressure in the high-pressure reactor are lowered to 100 °C and 10 bar, respectively, followed by the addition of 1-hexene to initiate the catalytic reaction. After 1 hour, a turnover number (TOs) of 68 is observed, which increases to 619 after 29 hours. The aldehyde L:B selectivity is 0.8, and the alkene isomerization rate is 15.1%, with no detectable production of alkanes or alcohols. This cationic Co(II) bisphosphine catalyst exhibits significantly higher activity compared to another recently reported inexpensive iron catalyst, H2Fe(CO)2(PPh3)2 (J. Am. Chem. Soc., 2018, 140, 4430–4439).
The authors also conducted catalytic reactions using 1-hexene and both a Co(I) model catalyst and PBu3 under industrial conditions to compare the cationic Co(II) bisphosphine catalyst with the neutral Co(I) catalyst system. The results demonstrated that the cationic Co(II)-DPPBz catalyst is at least 30 to 60 times faster than the typical neutral Co(I) catalyst system containing phosphine ligands.
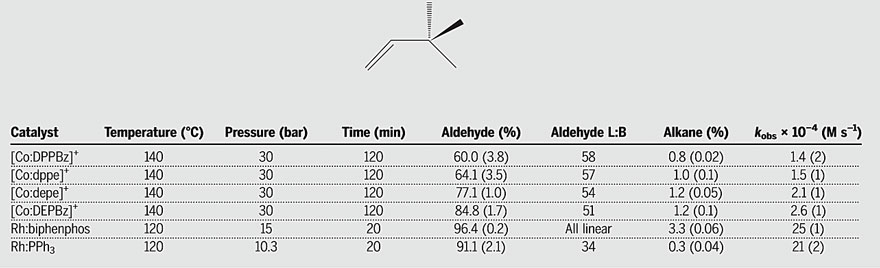
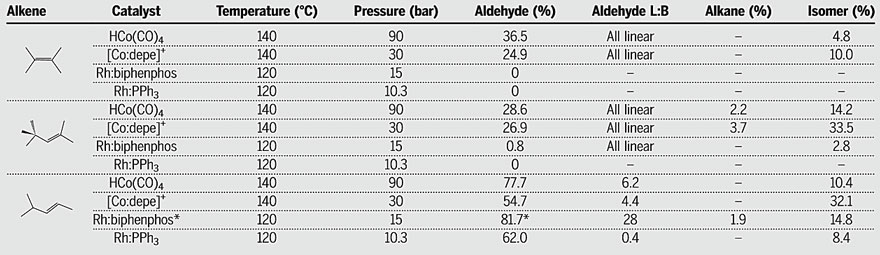
Next, the authors compared the
performance of four different cationic Co(II) bisphosphine catalysts and two
Rh-phosphine catalysts for the hydroformylation of 3,3-dimethylbutene (Figure
4). Among them, the Rh-biphenphos catalyst is known as one of the most active
and selective hydroformylation catalysts, but it is prone to undergo phosphite
decomposition, leading to shortened catalyst lifespan. Cationic cobalt
catalysts based on ethyl-substituted bisphosphine ligands (depe and DEPBz),
which are strong σ-donors, exhibit higher activity than cobalt catalysts based
on other bisphosphine ligands. Compared to rhodium catalysts, their observed
rate constants (kobs) can reach approximately one-tenth of that of the
rhodium catalysts. Considering that cobalt catalysts operate at higher
temperatures and pressures, the rate constants of these cobalt catalysts under
the same conditions might be approximately one-twentieth of that of the rhodium
catalysts. Although there is still a significant difference in activity,
considering that rhodium is at least 4000 times more expensive than cobalt, a
20-fold difference in activity seems acceptable.
Stability is crucial for
assessing the overall quality of a catalyst system. As an example,
[Co(acac)(depe)](BF4) showed excellent stability even after 336
hours (14 days) of reaction at 160 °C and 50 bar H2:CO (1:1)
conditions, with an average turnover frequency (TOF) of 59.5 min-1.
It is worth noting that the catalyst maintained good activity even after 336
hours, demonstrating its remarkable stability. The authors further investigated
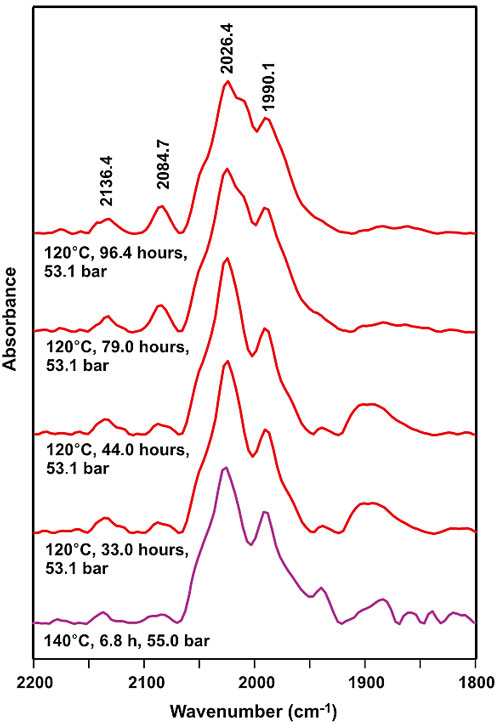
the catalytic process using in-situ Fourier Transform Infrared (FTIR)
spectroscopy (Figure 6), revealing that a 19-electron (19e-)
intermediate may play a crucial role in the catalysis. Finally, the authors
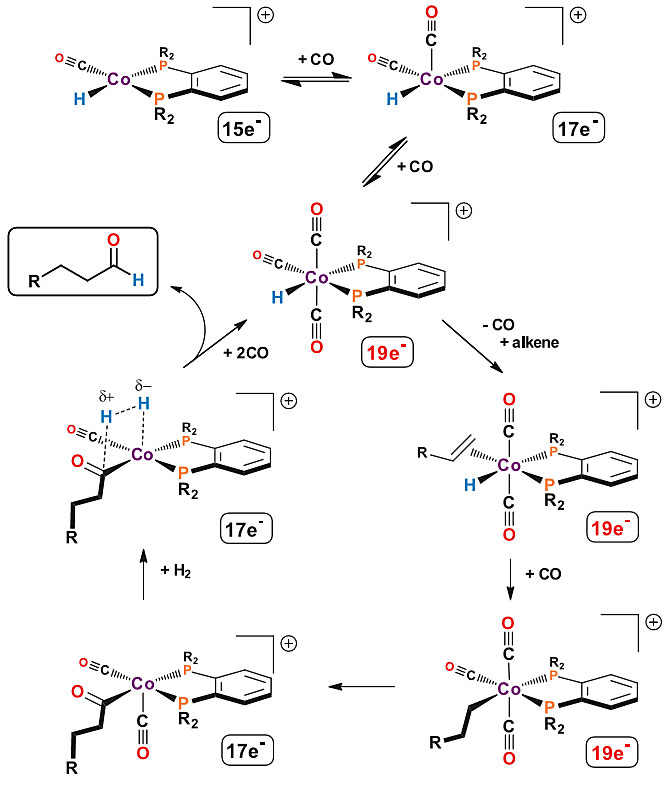
proposed a possible mechanism for this type of cationic Co(II) bisphosphine
catalyst (Figure 7). The basic reaction steps are similar to those of known
hydrogenation catalysts: olefin coordination, hydride migration insertion to
form alkyl intermediates, and CO and alkyl migration insertion to form acyl
species.
In the past 50 years, there
has been little research on cobalt-based hydrogenation catalysts, and there
have been few significant breakthroughs in the hydrogenation process as a
whole. This cationic Co(II) diphosphine hydrogenation catalyst reported by
Professor George Stanley's research group shows activity comparable to the
expensive rhodium catalysts currently used in industry. It does not require
high pressure or high temperature, exhibits high stability, and can achieve
hydrogenation of branched internal alkenes. With promising industrial
applications, it can be considered the most significant breakthrough in the
hydrogenation process in the past 50 years. Professor Stanley said, "The
best rhodium catalyst is only about 20 times faster than this cationic cobalt
diphosphine catalyst, but it is thousands of times more expensive"...
Rhodium catalysts generally require high temperatures and pressures, whereas
this cationic cobalt diphosphine catalyst can catalyze the reaction at moderate
pressure, making it a more energy-efficient technology. [1]
TEL:021-37896100
EMAIL:icti@shicti.com
ADDRESS:Building 11, No. 300, Dingyuan Road, Songjiang District, Shanghai

沪ICP备19009172号-3Copyright © 上海簇睿低碳能源技术有限公司 All Rights Reserved.